Catalytic Asymmetric Amination of Sulfenamides by DFT-guided Catalyst Design
2026-01-06 | 7 minstags: interactive, computational_chemistry, publication
This post reuses figures and tables from an ACS publication1. Reproduction follows the ACS guidelines.2
This blog post contains interactive visualization for the “Peptide-Catalyzed Asymmetric Amination of Sulfenamides Enabled by DFT-Guided Catalyst Optimization”1 by Tampellini, N.; Choi, E. S. and Miller, S. J.* (J. Am. Chem. Soc. 2025)1.
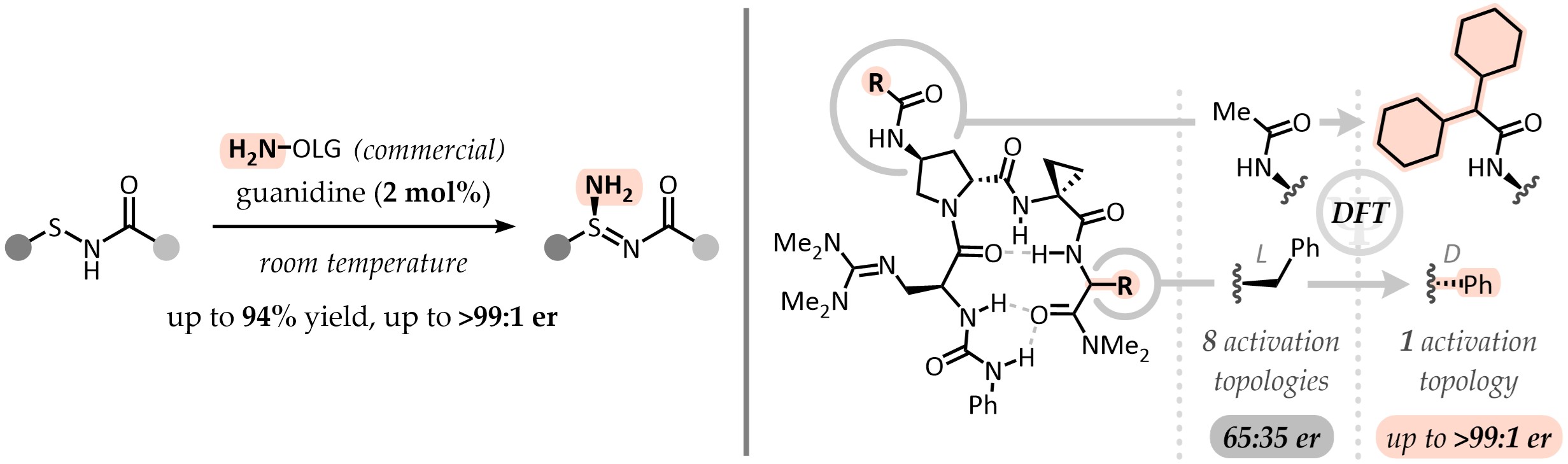
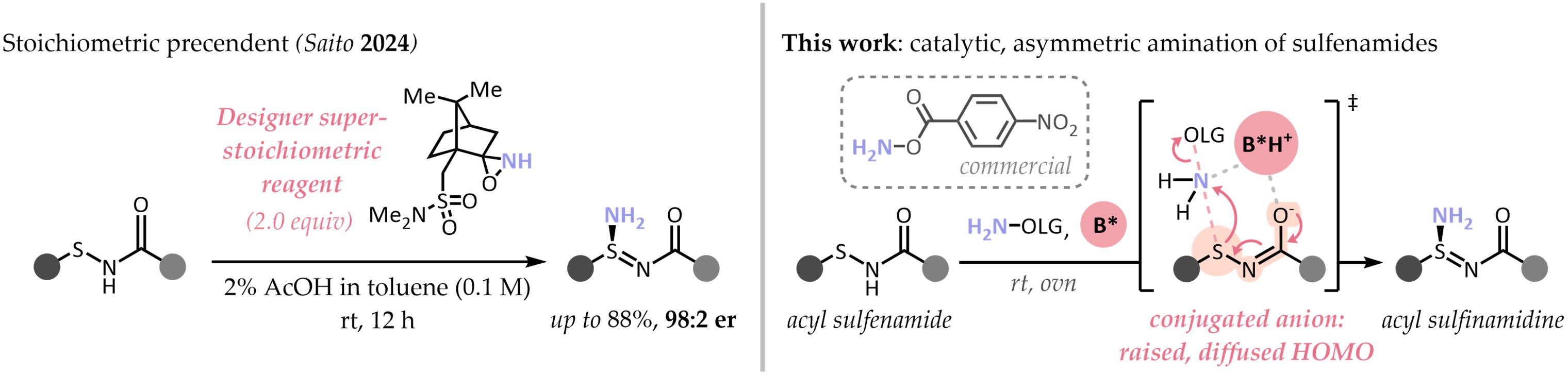
Recent times have seen a new interest in chiral-at-sulfur functional groups, but asymmetric methods to prepare them remain scarce. After a report from Saito and coworkers on the electrophilic amination of sulfenamides with a chiral reagent, we realized the lack of a catalytic, asymmetric approach to sulfinamidines. Instead of acidic activation, we reasoned that the reaction could proceed under base catalysis as well. The energetically raised, diffused HOMO of the sulfenamidate anion would therefore react with an electrophilic aminating reagent. Gladly, reactivity was straightforward to establish - but enantioselectivity proved more challenging.
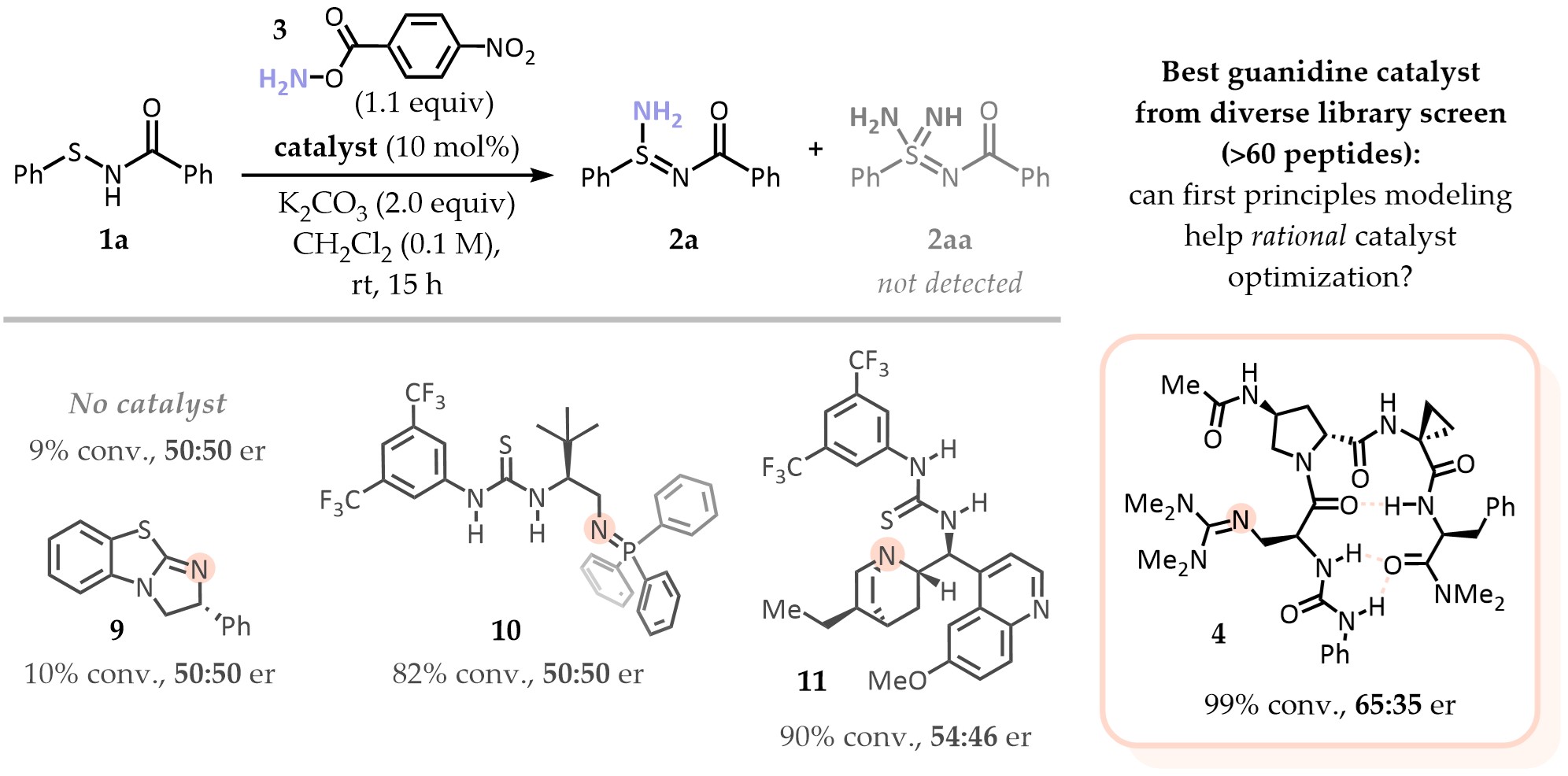
The screen of established chiral bases to direct the enantioselective amination was initially not very promising: only a few, mild hits emerged from a sensibly sized library. While peptidyl guanidines appeared as the most promising scaffold, our diverse library showed low variability in terms of enantioselectivity, with all catalysts performing similarly (~0-30% ee). We clearly didn't have the right catalysts for this transformation, and perhaps the community also did not. So how do we get to one that works? We challenged ourselves going for a DFT-first optimization approach, seeking to understand what lies beneath the mild (and unique) success of peptidyl guanidines as well as what doesn't work very well. From there, we can have a better idea of the catalyst chemical space we would need to explore.
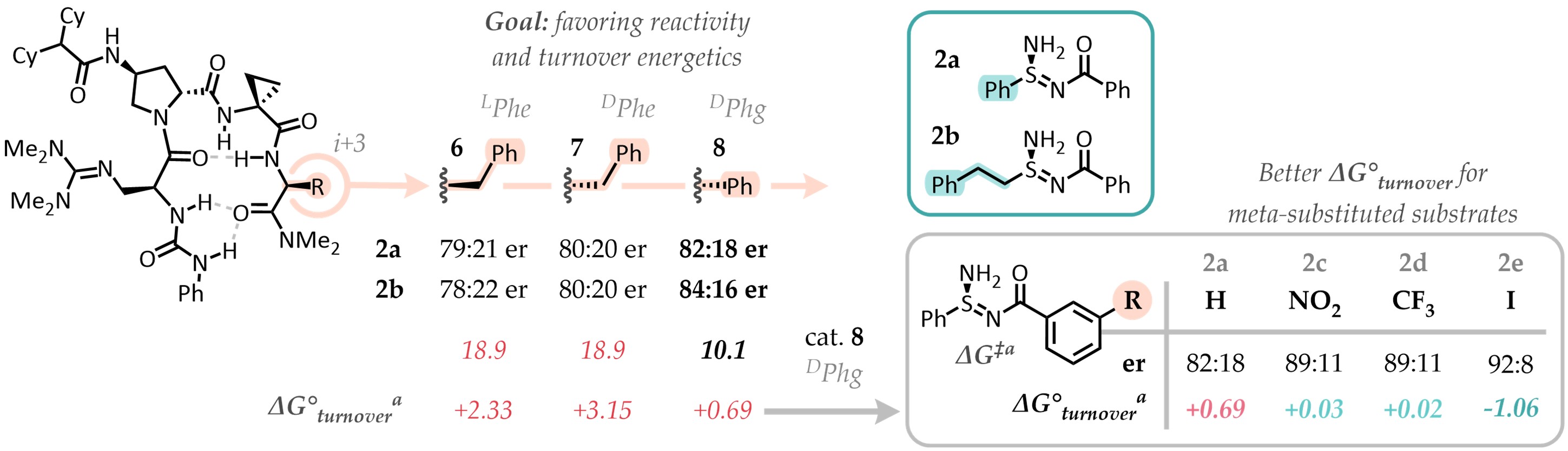
Hit catalyst 4, as expected, featured many energetically accessible activation modes - we targeted one as the target of our computational optimization. This mode positioned groups in a way that we believed could accomodate numerous substrates, while directing the amination reaction. Hence, we tested the strategic addition of steric bulk to a pendant amide position, with the idea of limiting the conformational space of the catalyst-substrate complex to that single mode. This would increase the ΔΔG‡ and ideally increase enantioselectivity. After just a few iterations, DFT calculations suggested that we had done it. Single activation mode, ΔΔG‡ > 4 kcal/mol! But wait, why is the selectivity still only about 4:1 er, then?
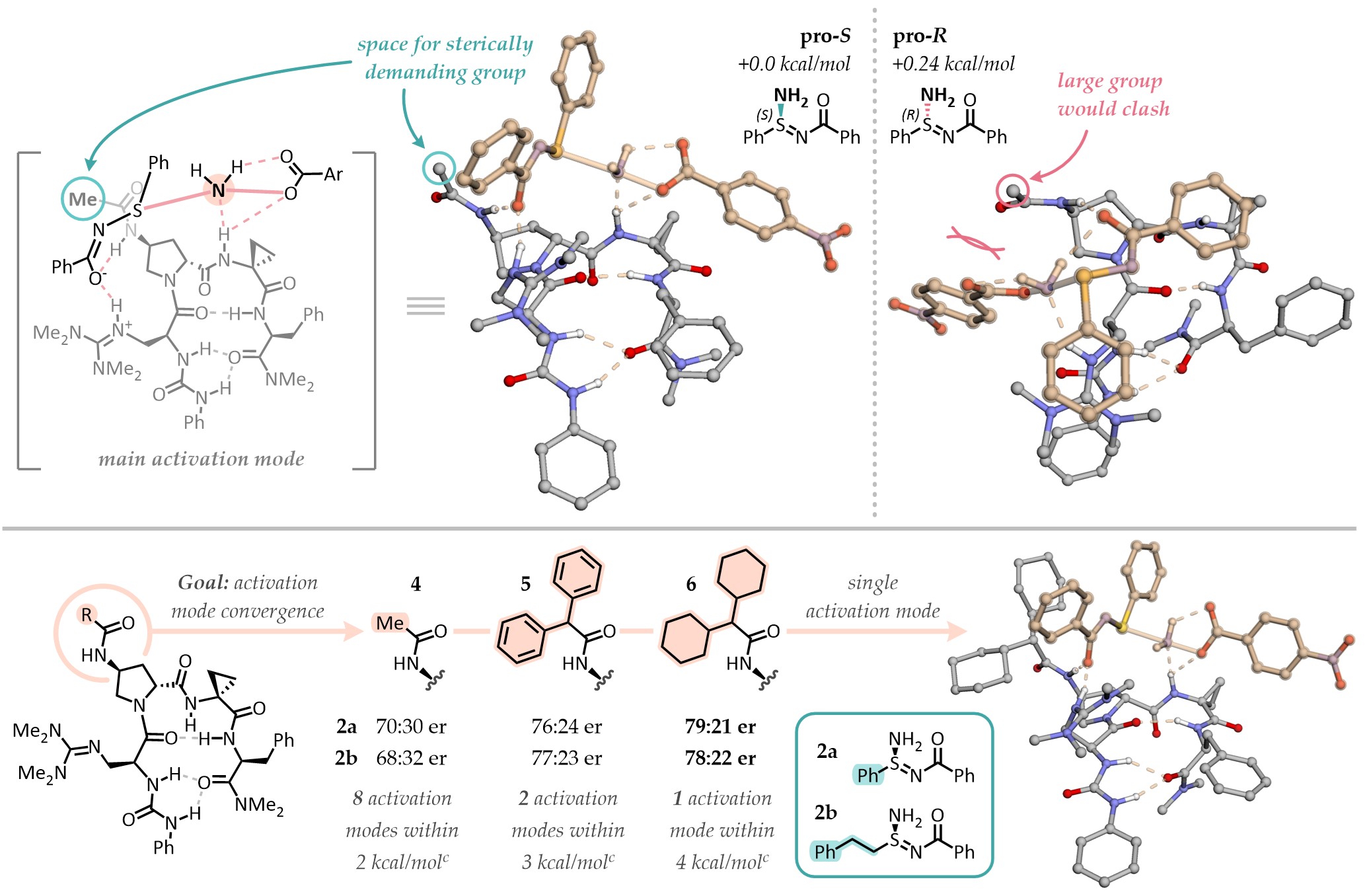
It turns out that we had done too much of a good job at designing the catalyst. It proved, in fact, to be such a good binder of the transition state that it was also binding the stereoelectronically similar products (post-reaction complex) too tightly. This is a problem if you are developing a catalytic reaction, where you need to displace the products from the catalyst to start a new cycle. DFT thermochemistry, in fact, informed us that catalyst turnover (exchanging the reaction products for the reagents) was endergonic for the substrates we were looking at. This meant that after some initial progress, the reaction products would inhibit the catalyst and other non-enantioselective processes would mediate the unselective formation of our chiral sulfinamidine products. The key to escape this issue was to find substrates that had an easier time dissociating from the catalyst. These turned out to be meta-substituted benzoyl sulfinamidines. While this seems a little specific, this moiety is easy to install and remove later in derivatizations - practically a protecting group!