Enantioselective S-Oxidation with an Aspartic acid-derived Peptide
2024-10-01 | 1 mintags: interactive, computational_chemistry, publication
This post reuses figures and tables from an ACS Catalysis publication1. Reproduction follows the ACS guidelines.2
This blog post contains interactive visualization for the computational section of our work on the enantioselective S-oxidation of sulfoxides by an aspartic-acid containing tetrapeptide.1
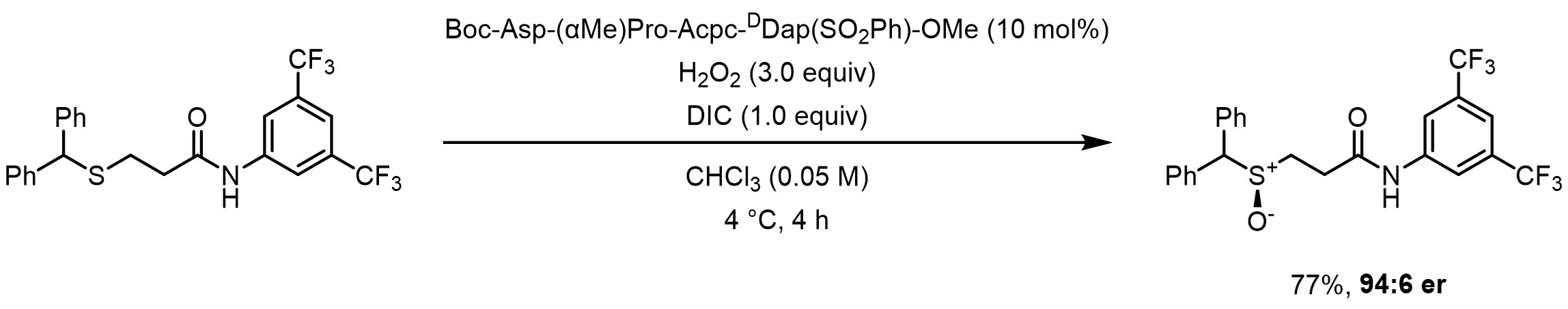
The modeled reaction.
Major product TS (pro-R)
Best transition state leading to the major enantiomer. Substrate highlighted in orange.
Minor product TS (pro-S)
Best transition state leading to the minor enantiomer. Substrate highlighted in orange.
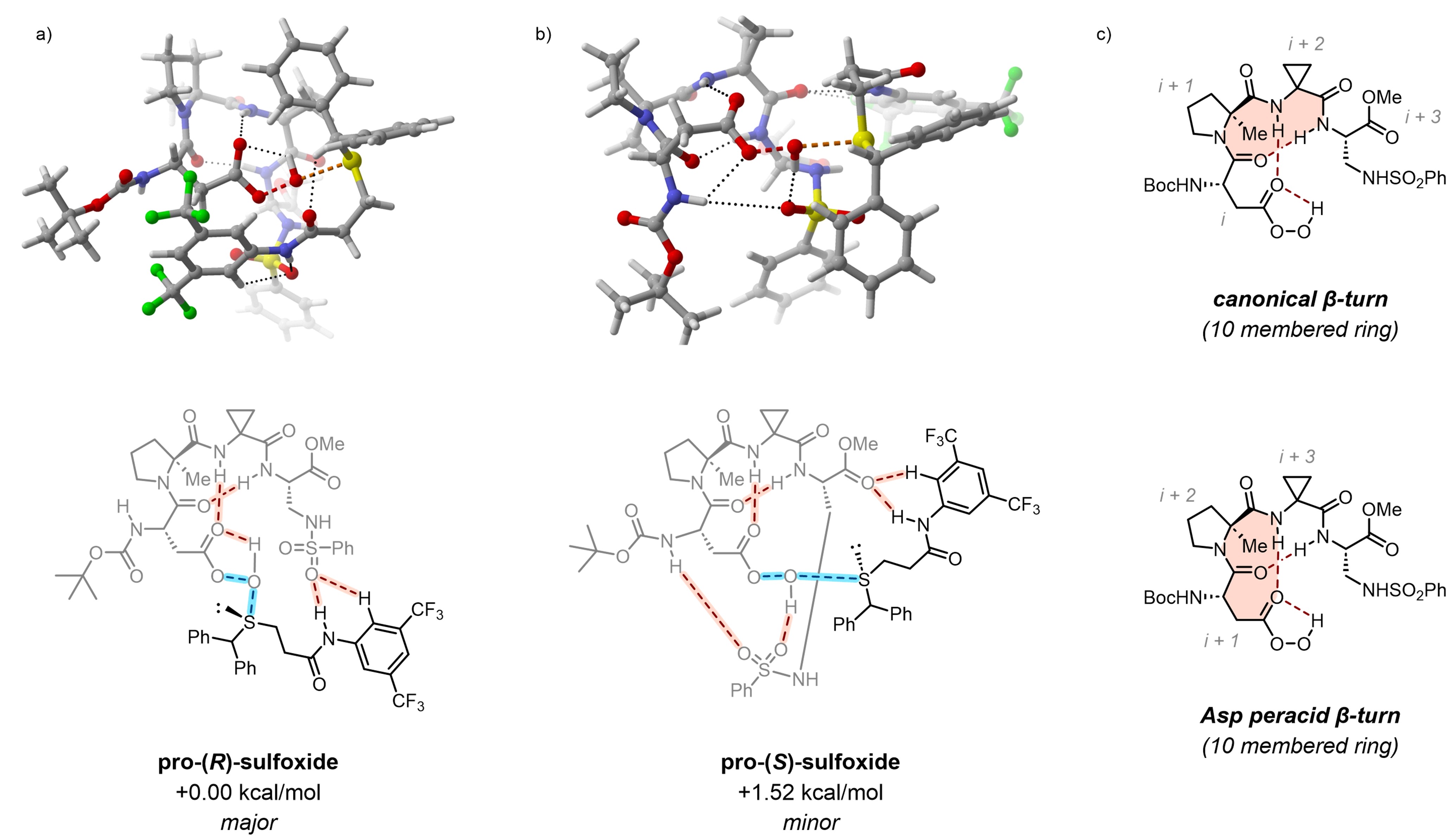
Two- and three-dimensional non-covalent binding network depictions. a: Major enantiomer best TS. b: Minor enantiomer best TS. c: Double β-hairpin motif within the catalyst. Energetic data at the ωB97M-V/def2-TZVPP/SMD(CHCl3)//R2SCAN-3c/CPCM(CHCl3) level.
-
The full paper is now published in Organic Letters. ↩ ↩2
-
See ACS guidelines for scholarly posting & sharing policies ↩